Alzheimer’s Disease and Apoptosis regulation are related to each other. This article explains the role of Apoptosis in causing Alzheimer’s disease.
DEFINITION OF APOPTOSIS:
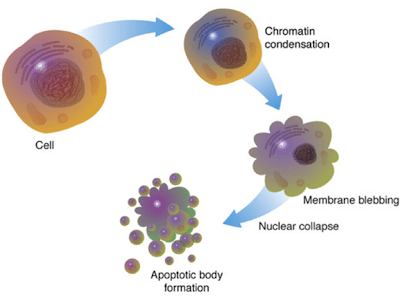
Apoptosis is the course of programmed cell death that occurs in eukaryotic organisms due to some biochemical events. These changes include forming a bulge of the plasma membrane of a cell, cellular contraction, DNA & nuclear fragmentation, chromatin condensation, and decaying of mRNA. It is used during early development to eliminate unwanted cells; for example, to develop the space between the fingers of a developing hand. In human adults, apoptosis is used to get free from the cellular body that have been damaged beyond repair. The average human adult loses between 50-70 billion cells per day due to apoptosis. For an average human child between the ages of 8 & 14, approximately loses 20–30 billion cells per day.

About the image: Long-term live cell imaging (12h) of multinucleated mouse pre-Adipocyte trying to undergo mitosis. Due to the excess of genetic material the cell fails to replicate and dies by apoptosis.
ROLE OF APOPTOSIS IN DISEASES:
Apoptosis plays a very important role within the regulation of the immune system. T-lymphocytes are cells formed from the the immune system which carries out the function of destroying infected or damaged cells in the human body. They develop in the thymus but they are checked while entering the bloodstream so that they are effective against foreign antigens and are also not reactive against healthy cells. Any ineffective or self-reactive T-cells are removed through the apoptosis induction. Regulatory difficulties of apoptosis have been implicated in many diseases. Cancer is a disease that is often characterized by scarce amount of apoptosis. Cancer cells typically possess several mutations that have allowed them to ignore normal cellular signals regulating their growth and become more proliferative than normal.
Under normal situation damaged cells will perform apoptosis, but if mutation occurs in apoptotic genes then it stops the cells from performing apoptosis. In such situations there is no scanning on the cellular proliferation and consequently increasing the population that leads to the formation of tumors. In several cases, it has been found that these tumors can be difficult to kill as many cancer treatments rely on damaging the cells with radiation or chemicals but the transformation or genetic mutation is in apoptotic pathway. Hence the function of apoptosis in cancer is therefore of keen interest among the scientists in the development of treatments for this disease. Not only small amount of apoptosis is detrimental but there are other assorted diseases where a large amount of apoptosis is thought to be a cause of the problem. For example in neurodegenerative diseases such as Parkinson’s or Alzheimer’s or Huntington’s diseases apoptosis is thought to account for much of the cell death and the progressive loss of neurons. So, here in this article I am going to write about one of such neurodegenerative disease, Alzheimer’s disease.
ALZHEIMER’S DISEASE:
Alzheimer’s disease (AD) is the most common type of dementia found among geriatric populations worldwide and is growing quickly in low- and middle-income countries. Alzheimer’s disease affects approximately 36.6 million people, and that number is expected to double over the next two decades. Those most susceptible to Alzheimer’s over the age of 60, though other associated factors such as sex, poor nutrition, education, impaired functional status, body mass index, diabetes, depression, smoking, alcohol, fish intake, and pesticide exposure have been reported, though none are clear.

Gaining a better understanding of the etiology of AD requires multiple-site-targeted therapy to control the disease at the initial level. On the other hand, evidence suggests that risk factors for AD are modifiable. Hence reduction in associated risk factors may require very long follow-ups to make people aware of their effect on AD incidence. If these factors are effective in preventing the progression of AD, the target populations could be affected at early stages in AD or even patients with more advanced disease.
BRIEF HISTORY OF ALZHEIMER’S DISEASE:

Described nearly a century ago by Alois Alzheimer in Alzheimer’s disease (AD) is characterized by an early deterioration in memory followed by progressive impairment of other cognitive domains and concurrent large-spectrum psychological and behavioral symptoms (1). Prior to the description of AD, the disease was primarily characterized as simple dementia. The mistake is natural, given that AD is the most common form of dementia in geriatric populations, accounts for 50 to 80% of all dementia cases (2). Impairments like dementia or AD have long been associated with advanced age. In roughly 2000 BC ago, ancient Egyptian seemed to aware that advanced age was associated with significant memory disorder (3), while over a millennia later, Solon wrote in his book that judgment could be impaired by physical pain, violence, drugs, and old age (4). Curiously there are some references to diseases like AD being distinct from dementia; for example, writers of Greek and Roman history presume the fabled stories of Agamemnon were true, but they identified psychopathology other than dementia. Unfortunately, these references disappeared by the middle ages—perhaps in an era marked by plague, religious warfare, and upheaval dementia did not inspire much interest or concern. Coincidentally, during this period, there were religious proscriptions against medical dissection. With the rise of the enlightenment and renewed interest in understanding the human body and mind (and a more open-minded attitude towards learning about either), there became renewed interest in understanding dementia. In the early 20th century, Alzheimer was investigating a 51-year-old woman who exhibited an unusual form of amnesia and an inability to answer questions and some other unusual symptoms. After her death, Alzheimer’s post-mortem examination of the body showed some novel features, notably distinctive plaques and tangles in the brain that we now know as neuritic plaques and neurofibrillary tangles. For a large part of the 20th century following this discovery, the only way to diagnose AD was to wait for the patient to die and examine the brain, and for most of this time, little Progress was made in actually understanding the etiology of the disease.
DIAGNOSTIC CRITERIA OF ALZHEIMER’S:
The development of diagnostics for AD was comparatively slow. For half a century after discovering the disease, the symptoms we now associate with Alzheimer’s were presumed to be part of the normal aging process. By the 1960s, there were some documented connections between cognitive decline and the presence of neuritic plaques and neurofibrillary tangles. This set the foundation for understanding AD as a complex disease with externally visible symptoms that could be diagnosed. By 1984, neurologists and pathologists had in place a series of guidelines to follow for diagnoses individual’s symptoms as assessed by family and friends and a basic neurological assessment (5). Unfortunately, AD manifests itself differently in each individual, making such assessments haphazard. There are, however, some common symptoms, including memory loss (forgetting an event, repeating sentences and statements, misplacing things or placing them to illogical locations, forgetting names of family members, etc.), disorientation and misinterpretation of spatial relationships, problems with speaking, writing, reasoning and concentration, changes in personality and behavior, depression, or difficulty performing normal daily tasks (6).
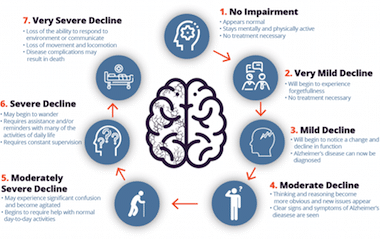
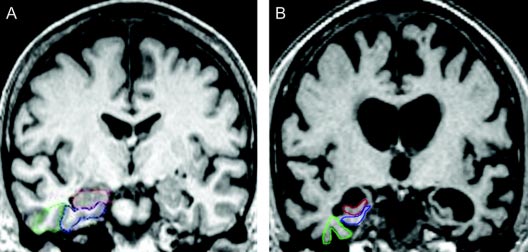
Recent advances in neurobiology and the pathogenesis of neurodegenerative diseases as well as the immense achievements of studies on human genetics paved the way for some updated diagnostic criteria and guidelines that were incorporated in 2011. The most important changes include the identification of three stages of Alzheimer’s disease before memory loss, and the search for Biomarkers test to confirm the presence and absence of the disease. These stages are mapped out to correspond to the severity of the disease. The first, Stage 1 (Mild) can last 2-4 years (7). During this period, patients with AD are less energetic and spontaneous, experience minor memory loss, exercise poor judgment, repeating sentences, and may withdraw, avoiding new places in favor of familiar ones. Next, Stage 2 (Moderate), the longest stage, lasting 2-10 years (8), is marked by the affected individual become clearly becomes disabled, requiring help in performing numerous tasks. Patients at Stage 2 may become more disconnected from their family and confused, forgetting recent events, family history, and even encounter difficulty recognizing familiar people. Similarly, speech problems increase, and patients may encounter problems in understanding reading, writing, and may invent new words. As the patients become aware of this loss of control, they may become dejected, short-tempered and restless. The final stage of the AD, Stage 3 (Severe) can last up to 3 years (9) during which patients encounter severe losses in basic functions, even losing their ability to feed themselves, speak, recognize people, and control bodily functions, (e.g., swallowing or bowel and bladder control). By this point, memory worsens and may become almost non-existent, patients sleep often and grunt or complaining. At this stage, constant care is critical. In a weakened physical state, patients may become susceptible to other illnesses such as skin infections or respiratory problems, particularly when they are unable to move on their own (10).
The second aspect of the new diagnostic criteria is that patients suspected of either having or being susceptible to having AD should undergo a biomarker test. While several studies have attempted to numerous potential biomarkers, the two accepted markers identified for Alzheimer’s disease include beta-amyloid and Tau proteins, and their level in blood and cerebrospinal fluid determine the disease at early stages (11).
PREVALENCE AND AGE FACTOR:
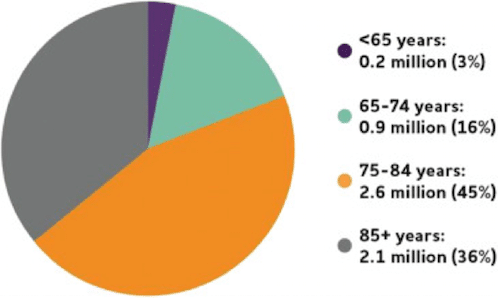
Current epidemiological forecasts predict that AD will have a marked impact on both the medical system and society at large due to the prevalence of AD dementia and the distribution of Alzheimer’s being most reported in patients 60-65 years old (12,13), which will continue to form a much larger share of the population in developing and developed countries worldwide. While early onset of Alzheimer’s can occur much earlier than 60 years of age, the frequency to dementia is relatively low at this early stage, and there is exponential increase in the dementia with age (reaching almost 50% those in 85 years of age). This risk increases even further past 85, from 12.5% per year after 90 years of age, 90-94 years age group to 21.2% per year in the 95-99 years old age group, and to 40.7% per year in those at least 100 years of old.
The age association is especially troubling in terms of medical care. The severe progressive nature of AD leading to worsening functional and cognitive decline and increased risk of the disease is becoming a major determining factor in the institutionalization of the elderly. This heterogeneity may result from differences in localized severity of brain damage or variations in patient’s personalities, life history, environmental risk factors and socio-economic conditions that may also be the cause of metabolic cognitive syndrome (MetS), which is also linked with the AD.
POTENTIAL CAUSES OF ALZHEIMER’S DISEASES:
An adult human brain contains 100 billion neurons and 100 trillion synapses which allow the signals to travel throughout the brain circuits and create thoughts, memories, skills and movement (14). In AD, interference causes disturbance in the function of these neurons and synapses, the number of synapses decline, neurons die and information transfer at synapses begins to fail. Precisely why this occurs is not clear. What we currently know is that AD is a chronic multi-factorial brain disorder that develops in response to multiple risk factors including environmental factors, chemical imbalances in the brain, hereditary factors and other brain development abnormalities. Studies on AD have noted a clear autosomal dominant pattern in people reported for AD before 65 years of age and people with inherited autosomal dominant genes who have not yet develop symptoms (15). Currently three genes mutations have been described that may lead to early forms of AD —amyloid precursor protein (APP), presenilin 1 (PS1) and presenilin 2 (PS2). Mutations in these genes seems to alter the proteolytic processing of amyloid precursor protein, leading to an increased production of the β-42, which forms the core of neuritic plaques and has been shown to act as a neurotoxin (16).

A broad range of environmental components may exacerbate damage to the brain and contribute to either the severity or risk of AD. For example, some environmental factors interact with the genetic liability in a negative manner to produce disorders. In terms of genetic risks, since mental illness can run in families, family history provides strong evidence that AD is more likely to develop in first degree relatives who have a parent, brother or sister with Alzheimer’s than those who do not have a first-degree relative with Alzheimer’s (17,18). Those who have more than one first-degree relative with Alzheimer’s are at even higher risk of developing the disease (19). Other studies show that illiterate people are at higher risk for AD than those who are highly educated (20). According to the cognitive hypothesis, additional years of education may increase the connection between neurons and activate the brain to compensate for the early brain changes of Alzheimer’s by using alternate routes of neuron-to-neuron communication to complete a cognitive task (21,22). Other studies suggest that other modifiable factors, such as remaining mentally (23) and socially active, may support brain health and possibly reduce the risk of Alzheimer’s and other dementias (24,25), though further follow-up studies are needed to verify these possibilities.
TREATMENT METHODS TO CURE ALZHEIMER’S DISEASES:
Source: Health Central
In brief, during AD neurons are chief cells being destroyed. In the brain, neurons connect and communicate at the synapse, where tiny bursts of neurotransmitters carry information from one cell to another. AD disrupts this process, eventually destroying synapses and killing neurons, thereby damaging the brain’s communication network and leading to the gradual degradations in cognitive and motor function. Curing the disease would then necessitate first stopping the process that leads to the death of neuron cells, and then finding a way to actually regrow damaged or dead neurons. The potential difficulty in this venture is enormous. No wonder that at the moment there are no specific strategies that can fully cure the disease. Instead, there are three potential venues that if pursued correctly may help to inhibit the disease’s progression:
- Prevention. To prevent the onset of AD, there are several possibilities: Direct genetic manipulation; Inhibition of β-amyloid production; or Inhibition of β-amyloid aggregation. Currently, the main drug used for this purpose is Cholinesterase, which works by slowing down the disease activity that breaks down key neurotransmitters, i.e. Donepezil, galantamine, rivastigmine,and tacrine.
2. Arresting Progress. Given the nature of AD, the disease’s Progress can be arrested by using anti-inflammatory drugs, antioxidants / free radical scavengers, and other neuroprotective methods. Arresting Progress seems to be quite promising and moderately successful in that it can delay the risk and time of the disease.
3. Symptomatic Treatment. Since we neither know enough of the underlying causes of AD to develop a targeted treatment nor have the technical capacity to actually reverse the damage caused by the disease, the extant treatments focuses on the replacement of neurotransmitter (acetylcholine, glutamate and serotonin) losses during the course of AD. Cholinesterase inhibitors have proven a successful approach in delaying cognitive deterioration, and serotonergic system can be manipulated by the use of atypical antipsychotics have proven a viable strategy for the treatment of some non-cognitive symptoms, but again until the bases and risks for AD are more clearly understood, these are merely palliative care, and not an actual solution.
SUMMARY AND FUTURE DIRECTION:
Alzheimer’s has a troublingly double-edged sword. On one hand, we do not clearly understand it or how to cure it, and on the other, for reasons unknown incidences of AD keep growing. Accordingly, researchers are constantly looking for new ways to treat the disease. At the moment, a promising focus is working on pre-symptomatic Alzheimer’s subjects (carrying genetic determinants which eventually will develop in to Alzheimer’s) or asymptomatic subjects (which are at a risk of AD with biomarkers of AD pathology). Working with these populations informs how to avoid the risks before they develop further, theoretically offering novel opportunities for new treatment options. Other research groups are also working on non human primate Alzheimer models and to try and understand the underlying nature of AD the efficacy of novel treatment options.
Now, there are so many things that we have to keep in mind while exploring this factor. Researcher’s need to fins the missing links in all this process. If we focus on the dietary habitat then the history of the AD began 20 year or so ago, the time when the trend of coca cola and diet soft drinks started. These drinks contains high amount of formaldehyde. May be a case repot will help us to solve it. There are some studies in mice and rats, which show the formaldehyde do not have any effect, and accumulation of beta amyloid plaque were not observed. But, more research is needed to study the effect of formaldehyde in non-human primates and resolve such issues.
REFERENCES:
- Berrios GE. Alzheimer’s disease: a conceptual history. International Journal of Geriatric Psychiatry 1990; 5: 355-365.
- Signoret JL, Hauw JJ. Alzheimer’s disease and other dementias. 1re ed. Medicine Science Flammarion, Paris, 1991; 511.
- Freeman K. The work and life of Solon. London: London University. 1926; Press.
- Berrios GE. Non-cognitive symptoms and the diagnosis of dementia. Historical and clinical aspects. Br J Psychiatry Suppl.1989; 4: 11-6.
- Russell A, Barkley Kevin R, Murphy . Attention-deficit Hyperactivity Disorder: A Clinical Workbook. 2: chap. 8: 337. 2006.
- Robinson P, Giorgi B, Sirkka-Liisa E. The Lived Experience of Early-Stage Alzheimer’s disease: A Three-Year Longitudinal Phenomenological Case Study. J. Phenomenol. Psych 2012: 43: 216-238.
- Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM, Fagan AM. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013; 12: 957-65.
- Hellweg, R, Wirth Y, Janetzky W, Hartmann S. Efficacy of memantine in delaying clinical worsening in Alzheimer’s disease (AD): responder analyses of nine clinical trials with patients with moderate to severe AD. Int J Geriatr Psychiatry 2012; 27: 651-6.
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R et al., . The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement 2011; 7: 263-9.
- Ulrich J . Alzheimer changes in nondemented patients younger than sixty-five: possible early stages of Alzheimer’s disease and senile dementia of Alzheimer type. Ann Neurol 1985; 17: 273-7.
- Humpel C . Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol 2011; 29: 26-32.
- Fratiglioni L, Winblad B, Strauss E. Prevention of Alzheimer’s disease and dementia. Major findings from the Kungsholmen Project. Physiology & Behavior 2007; 92: 98–104.
- Winter Y, Korchounov A, Zhukova TV, Bertschi NE). Depression in elderly patients with Alzheimer dementia or vascular dementia and its influence on their quality of life. J Neurosci Rural Pract 2011; 2: 27-32.
- Jorm AF, Dear KB, Burgess NM. Projections of future numbers of dementia cases inAustralia with and without prevention. [Comparative Study]. The Australian and New Zealand journal of psychiatry 2005; 39: 959–963.
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob FW, Lent R, Herculano-Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 2009; 513: 532-41.
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ et al.,. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7: 280-92.
- Mayeux R, Sano M, Chen J, Tatemichi T, Stern Y.Risk of dementia in first-degree relatives of patients with Alzheimer’s disease and related disorders. Arch Neurol 1991; 48: 269–73.
- Green RC, Cupples LA, Go R, Benke KS, Edeki T, Griffith PA et al.,. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002; 287:329–36.
- Fratiglioni L, Ahlbom A, Viitanen M, Winblad B. Risk factors for late-onset Alzheimer’s disease: A population-based, case control study. Ann Neurol 1993; 33: 258–66.
- Lautenschlager NT, Cupples LA, Rao VS, Auerbach SA, Becker R, Burke J et al. . Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE Study: What is in store for the oldest old? Neurology 1996; 46: 641–50.
- Fitzpatrick AL, Kuller LH, Ives DG, Lopez OL, Jagust W, Breitner JC, et al.,. Incidence and prevalence of dementia in the Cardiovascular Health Study. J Am Geriatr Soc 2004; 52:195–204.
- Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD et al. . Dementia and Alzheimer disease incidence: A prospective cohort study. Arch Neurol 2002; 59: 1737–46.
- Evans DA, Bennett DA, Wilson, RS, Bienias JL., Morris MC, Scherr PA, et al.,. Incidence of Alzheimer disease in a biracial urban community: Relation to apolipoprotein E allele status. Arch Neurol 2003; 60: 185–9.
- Hall CB, Lipton RB, Sliwinski M, Katz MJ, Derby CA, Verghese J . Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology 2009; 73: 356–61.
- Wang HX, Xu W, Pei JJ . Leisure activities, cognition and dementia. BBA–Mol Basis Dis. 2012; 1822: 482–91.
Souhrid Sarkar
Souhrid Sarkar is an Aspiring Biotechnologist. He is a B.Tech student at Amity University Kolkata. He is content writer at BioXone. He is a Research Intern at Indian Institute of Technology, Bombay (IITB) & Reviewer at TMR Publishing Group. He is Keyboardist by hobby.


One thought on “Apoptosis Dysregulation and Alzheimer’s Disease”